# Attach libraries and functions:library(tidyGenR)library(tidyverse)library(ShortRead)source("code/gen2hierfstat.R")# conditionals to run chunksp_subseqs <-"output/variants_subseqs.rds"eval_subsample <-!file.exists(p_subseqs)if(file.exists(p_subseqs)) variants_subseqs <-readRDS(p_subseqs)
Effect of coverage on genotypes called
The read depth for each locus can have an effect on the reliability of the genotypes. Simulated lower coverage can be obtained by re-sampling lower number of reads from the FASTQ files. Lower coverage FASTQ datasets were created and variants were called with tidyGenR. Some statistics relevant to the performance of the genotype calls were tracked along decreasing coverage.
Locus selection
Select loci whose average number of reads across samples is above 500.
# compute reads per locustr_reads <-reads_loci_samples(trunc_dir, pattern_fq ="F_filt.fastq.gz" ) |>pivot_longer(-sample,names_to ="locus",values_to ="reads") |>group_by(locus) |>reframe(av_reads =mean(reads),sd_reads =sd(reads)) |>arrange(av_reads) |>filter(locus !="fetub") # likely amplifying multiple loci (see Camacho-Sanchez et al. 2026)# print loci to keeploci2keep <-filter(tr_reads, av_reads > thr_keep) |>pull(locus)print(loci2keep)
For the selected loci, a given number of reads will be randomly sub-sampled from the FASTQ.
Code
# input fastqfq <-list.files(trunc_dir, pattern ="fastq.gz", full.names = T)# sample sizessample_size <-c(500, 250, 125, 70, 50, 35, 20)# fix omega as in Camacho-Sanchez et al. 2026omega_a <-10^-2
Sample read subsets and call variants
For each category of read, 500, 250, 125, 70, 50, 35, 20, the following steps were performed:
For each FASTQ file:
Determine the number of reads in the FASTQ.
If the number of reads is below sampling_size, copy the FASTQ to a temporary directory.
If the number of reads exceeds sampling_size, randomly sample sampling_size reads and write the FASTQ to a temporary directory. A seed is used to maintain F/R pairing.
Code
variants_subseqs <- sample_size |>lapply(function(n_thr) {# create temp dir specific to sample_size sub_reads <-file.path(tempdir(), paste0("subsample", n_thr))if (dir.exists(sub_reads)) unlink(sub_reads, recursive =TRUE, force =TRUE)dir.create(sub_reads)# write subsampled fastq to temp dir fq |>lapply(function(fq_i) { n_reads <-countFastq(fq_i) out_fq <-file.path(sub_reads, basename(fq_i))if(n_reads$records < n_thr) {file.copy(fq_i, out_fq) } elseif (n_reads$records >= n_thr){set.seed(1) # critical for not loosing paired-read match sidx <-FastqSampler(fq_i, n_thr) sub_fq <-yield(sidx)writeFastq(sub_fq, out_fq,compress =TRUE)#try(close(sidx), silent = TRUE) # remove warnings }invisible(NULL) })# clean close conections open by ShortReadgc()# call variantsvariant_call(in_dir = sub_reads,loci = loci2keep,rv_pattern ="R_filt.fastq.gz",c_unmerged =TRUE,multithread =TRUE,pool =FALSE,omega_a_f = omega_a,omega_a_r = omega_a,band_size =0,ad =2,maf =0.1) }) |>setNames(paste0("sub_sample", str_pad(sample_size, width =3, side ="left", pad ="0")))saveRDS(variants_subseqs, "output/variants_subseqs.rds")
A short script is run to verify the pairing between F/R reads is kept.
Code
temp_dir_eg <-file.path(tempdir(), paste0("subsample", sample_size[length(sample_size)])) |>list.files(full.names = T)fq_ids <-lapply(c(1, 2), function(x) {readFastq(temp_dir_eg[x]) |>id() |>as.character() })if(identical(fq_ids[1], fq_ids[2])) {message("The pairing between F and R reads has been kept")} else {stop("The pairing between F/R paired reads has been lost.")}
Evaluate the effect on the calls
A first quick look at Figure 1 shows that genotype calls are consistent from 500 reads down to around 50. Below 50, but mostly at 20x, many alleles are not detected:
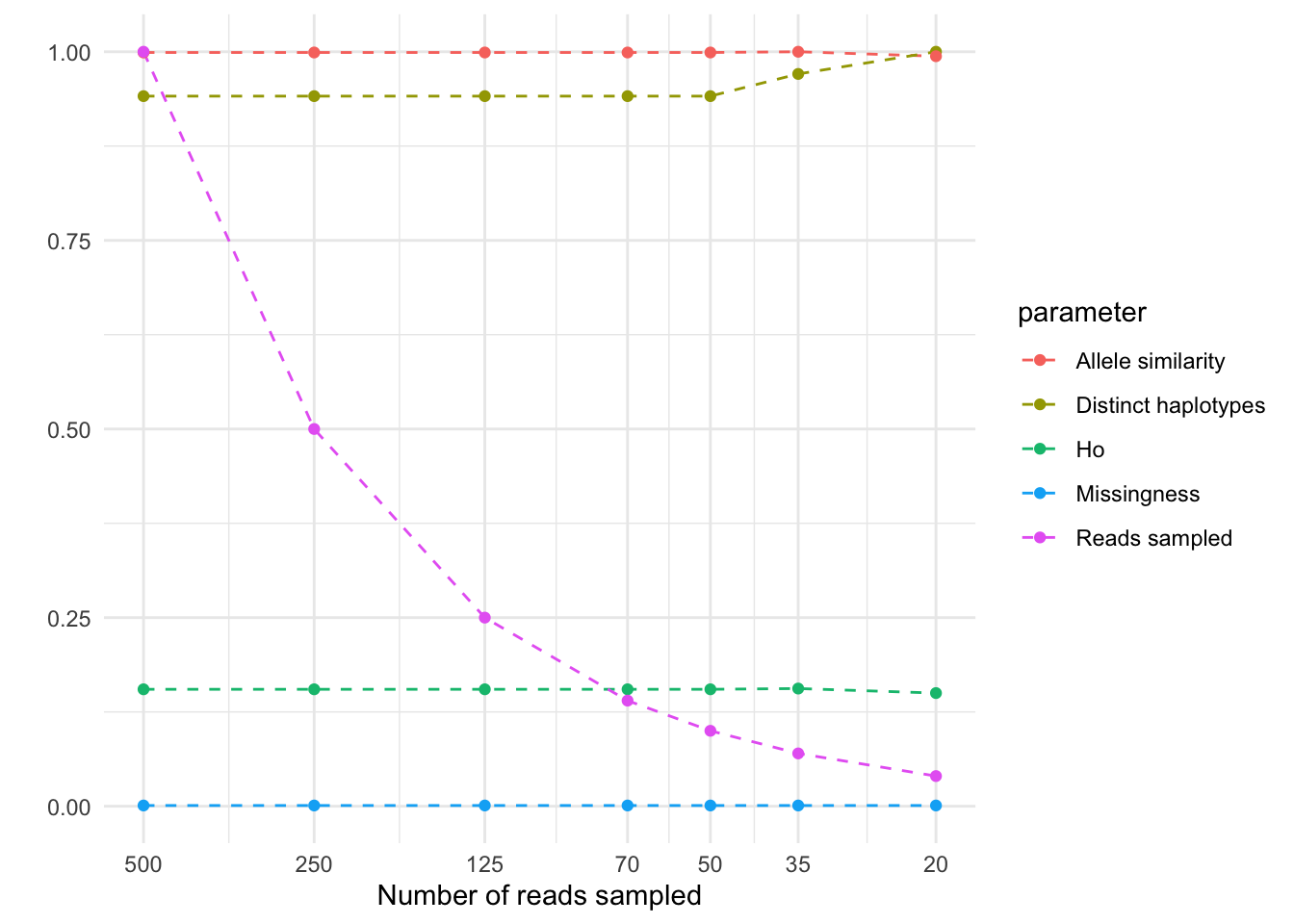
Figure 2: Assessment of quality parameters decreasing coverage.
Genotype calls were stable from 500x to around 50x (Figure 2). Below 50x, but mostly at 20x the missing calls increase from nearly 0 to 4%. The number of distinct haplotypes detected was little affected even in the lowest coverage (20x) tested. At 20x, some alleles from heterozygous genotypes were lost (see ‘Allele similarity’), causing the heterozygosity to sink from 35x to 20x.
Code
# save plotssaveRDS(list(p_stats = p_stats,p_comp = comp_gen$plot2),"output/plots-assessment-coverage.rds")
---author: Miguel Camacho Sáncheztitle: "Effect of coverage"---```{r}#| echo: false#| include: false#knitr::opts_knit$set(root.dir = "../")``````{r}#| message: false#| warning: false#| eval: true#| code-fold: true# Attach libraries and functions:library(tidyGenR)library(tidyverse)library(ShortRead)source("code/gen2hierfstat.R")# conditionals to run chunksp_subseqs <-"output/variants_subseqs.rds"eval_subsample <-!file.exists(p_subseqs)if(file.exists(p_subseqs)) variants_subseqs <-readRDS(p_subseqs)```## Effect of coverage on genotypes calledThe read depth for each locus can have an effect on the reliability of the genotypes. Simulated lower coverage can be obtained by re-sampling lower number of reads from the FASTQ files. Lower coverage FASTQ datasets were created and variants were called with *tidyGenR*. Some statistics relevant to the performance of the genotype calls were tracked along decreasing coverage.```{r}#| code-fold: true#| eval: true#| echo: falsetrunc_dir <-"data/intermediate/truncated"# minimum number of average reads for a locus to be keptthr_keep <-500```### Locus selectionSelect loci whose average number of reads across samples is above `r thr_keep`.```{r}#| message: false# compute reads per locustr_reads <-reads_loci_samples(trunc_dir, pattern_fq ="F_filt.fastq.gz" ) |>pivot_longer(-sample,names_to ="locus",values_to ="reads") |>group_by(locus) |>reframe(av_reads =mean(reads),sd_reads =sd(reads)) |>arrange(av_reads) |>filter(locus !="fetub") # likely amplifying multiple loci (see Camacho-Sanchez et al. 2026)# print loci to keeploci2keep <-filter(tr_reads, av_reads > thr_keep) |>pull(locus)print(loci2keep)```For the selected loci, a given number of reads will be randomly sub-sampled from the FASTQ.```{r}#| code-fold: true# input fastqfq <-list.files(trunc_dir, pattern ="fastq.gz", full.names = T)# sample sizessample_size <-c(500, 250, 125, 70, 50, 35, 20)# fix omega as in Camacho-Sanchez et al. 2026omega_a <-10^-2```### Sample read subsets and call variantsFor each category of read, `r sample_size`, the following steps were performed:- For each FASTQ file: - Determine the number of reads in the FASTQ. - If the number of reads is below `sampling_size`, copy the FASTQ to a temporary directory. - If the number of reads exceeds `sampling_size`, randomly sample `sampling_size` reads and write the FASTQ to a temporary directory. A `seed` is used to maintain F/R pairing.```{r}#| warning: false#| message: false#| eval: !expr eval_subsample#| echo: true#| results: hide#| code-fold: truevariants_subseqs <- sample_size |>lapply(function(n_thr) {# create temp dir specific to sample_size sub_reads <-file.path(tempdir(), paste0("subsample", n_thr))if (dir.exists(sub_reads)) unlink(sub_reads, recursive =TRUE, force =TRUE)dir.create(sub_reads)# write subsampled fastq to temp dir fq |>lapply(function(fq_i) { n_reads <-countFastq(fq_i) out_fq <-file.path(sub_reads, basename(fq_i))if(n_reads$records < n_thr) {file.copy(fq_i, out_fq) } elseif (n_reads$records >= n_thr){set.seed(1) # critical for not loosing paired-read match sidx <-FastqSampler(fq_i, n_thr) sub_fq <-yield(sidx)writeFastq(sub_fq, out_fq,compress =TRUE)#try(close(sidx), silent = TRUE) # remove warnings }invisible(NULL) })# clean close conections open by ShortReadgc()# call variantsvariant_call(in_dir = sub_reads,loci = loci2keep,rv_pattern ="R_filt.fastq.gz",c_unmerged =TRUE,multithread =TRUE,pool =FALSE,omega_a_f = omega_a,omega_a_r = omega_a,band_size =0,ad =2,maf =0.1) }) |>setNames(paste0("sub_sample", str_pad(sample_size, width =3, side ="left", pad ="0")))saveRDS(variants_subseqs, "output/variants_subseqs.rds")```A short script is run to verify the pairing between F/R reads is kept.```{r}#| code-fold: true#| eval: !expr eval_subsampletemp_dir_eg <-file.path(tempdir(), paste0("subsample", sample_size[length(sample_size)])) |>list.files(full.names = T)fq_ids <-lapply(c(1, 2), function(x) {readFastq(temp_dir_eg[x]) |>id() |>as.character() })if(identical(fq_ids[1], fq_ids[2])) {message("The pairing between F and R reads has been kept")} else {stop("The pairing between F/R paired reads has been lost.")}```## Evaluate the effect on the callsA first quick look at @fig-compare-calls shows that genotype calls are consistent from 500 reads down to around 50. Below 50, but mostly at 20x, many alleles are not detected:```{r}#| eval: true#| code-fold: true#| message: false# get genotypesgen_subseqs <- variants_subseqs |>lapply(function(x) genotype(x, ploidy =2, ADt =5))# compare callscomp_gen <-compare_calls(gen_subseqs)comp_vars <-compare_calls(variants_subseqs)# compare genotype vs variantscomp_gen_vars <-names(variants_subseqs) |>lapply(function(set_i) {compare_calls(list(var = variants_subseqs[[set_i]],gen = gen_subseqs[[set_i]])) }) |>setNames(names(variants_subseqs))``````{r}#| label: fig-compare-calls#| warnings: false#| fig-cap: "Comparisson of genotype calls with decreasing coverage."#| code-fold: true#| fig-width: 14#| fig-height: 9#| output-div: scroll-xcomp_gen$plot2```The genotypes from the subsets with larger number or reads are similar to each other.To summarise the results, the following statistics were computed:- **Observed heterozygosity** (*Ho*), since lower coverage could hinder the determination of both alleles, leading to an increase of homozygosity.- **Missingness**, measured as missing genotype calls (i.e. cells in the *sample x locus* matrix with no data).- **Allele similarity**, quantified as the average proportion alleles in each call that are identical to the best set (with more alleles genotyped).- **Distinct haplotypes**, or total number of distinct alleles considering all loci (expressed as a proportion respect the the maximum).- **Reads sampled**, reads sampled `r sample_size`, expressed as proportions to `max sample_size`.```{r}#| eval: true#| code-fold: true#| warning: false#| message: falserequire(hierfstat); require(scales)# total number of different haplotypes (sum across loci)no_variants <- gen_subseqs |>vapply(function(var_i) length(unique(var_i$md5)), FUN.VALUE =numeric(1L)) |>as.data.frame() |>setNames("distinct_hapl") |> tibble::rownames_to_column("subsample")# observed heterozygosityho <- gen_subseqs |>sapply(function(gen_i) { hf_i <- gen_i |>mutate(population ="trusmadi") |>gen_tidy2hierfstat() |>select(-sample, -population) |>basic.stats() ho_i <-round(hf_i$overall[which(names(hf_i$overall) =="Ho")], 3)# }) |>as.data.frame() |>setNames("Ho") |> tibble::rownames_to_column("subsample") |>mutate(subsample =str_remove(subsample, ".Ho$"))# calls with at least one haplotypecalls_plus_hap <- gen_subseqs |>vapply(function(gen_i) { gen_i_wide <- gen_i |>gen_tidy2wide() |>select(-sample)round(sum(is.na(gen_i_wide)) / (ncol(gen_i_wide) *nrow(gen_i_wide)),3) },numeric(1)) |>as.data.frame() |>setNames("missingness") |> tibble::rownames_to_column("subsample")# completnesscompletness <- comp_gen$tidy_dat |>select(-c(sample, locus, md5)) |>apply(2, sum) |>as.data.frame() |>setNames("allele_similarity") |> tibble::rownames_to_column("subsample")# create df for plotting how some stats change with coveragedf_fortified_abs <-left_join(no_variants, ho, by ="subsample") |>left_join(completness) |>left_join(calls_plus_hap) |>mutate(sample_size =str_extract(subsample, "[0-9]+") |>as.numeric())df_fortified_rel <- df_fortified_abs |>mutate(sample_size = sample_size /max(sample_size),distinct_hapl = distinct_hapl /max(distinct_hapl),allele_similarity = allele_similarity /max(allele_similarity) ) |>pivot_longer(-subsample, names_to ="parameter", values_to ="value") |>mutate(sample_size =str_extract(subsample, "[0-9]+") |>as.numeric())# plot stats p_stats <-ggplot(df_fortified_rel, aes(x = sample_size, y = value, colour = parameter)) +geom_line(linetype ="dashed") +geom_point() +scale_x_continuous(trans = scales::trans_new(name ="revlog2",transform =function(x) -log2(x),inverse =function(x) 2^(-x) ),breaks =sort(unique(df_fortified_rel$sample_size)),labels =sort(unique(df_fortified_rel$sample_size)) ) +scale_color_discrete(labels =c("Ho"="Ho","distinct_hapl"="Distinct haplotypes","allele_similarity"="Allele similarity","sample_size"="Reads sampled","missingness"="Missingness" )) +xlab("Number of reads sampled") +ylab("") +theme_minimal()knitr::kable(df_fortified_abs)``````{r}#| label: fig-assessment#| warnings: false#| fig-cap: "Assessment of quality parameters decreasing coverage."#| code-fold: true#| fig-width: 7#| fig-height: 5print(p_stats)```Genotype calls were stable from 500x to around 50x (@fig-assessment). Below 50x, but mostly at 20x the missing calls increase from nearly 0 to 4%. The number of distinct haplotypes detected was little affected even in the lowest coverage (20x) tested. At 20x, some alleles from heterozygous genotypes were lost (see 'Allele similarity'), causing the heterozygosity to sink from 35x to 20x.```{r}#| eval: true#| code-fold: true#| warning: false#| message: false# save plotssaveRDS(list(p_stats = p_stats,p_comp = comp_gen$plot2),"output/plots-assessment-coverage.rds")```# Session Info```{r}sessionInfo()```